GenBank
That is taken directly from Wikipedia today. There are billions and billions of nucleotides in there; probably billions of individual sequences and sequence fragments, plus all sorts of other data. So the question is, how did so much data get uploaded to Genbank when the main data entry portal to Genbank - the program Sequin - is so clunky and flawed?
Current options for uploading data include Sequin, which the last time I used it kept shifting coding sequences around on the mitochondrial genomes I was uploading; tbl2asn which requires that you are familiar with shell scripting, able to generate a tab-delimited data file that has a unique format almost impossible to generate automatically from the spreadsheet or .csv output from other programs; or Geneious, which so far has failed me in that it changes the genetic code from what is annotated in the software.
Yet somehow all these data are there. Most of them missing critical meta-data, like the latitude/longitude from which the sequence came, or who identified the specimen. That takes extra work, and NCBI doesn’t make it easy.
So how can we make this easier? For single-gene submissions, tbl2asn can be very easy because you can annotate most of the data as part of a FASTA file. But we are moving beyond the world of single-gene submissions very quickly. The complication of exons, introns, reverse-strand coded sequences, whole chromosomes, whole genomes means we all need to get more savvy about how to do this.
I don’t have the answer, I’m just complaining. I’m pretty computer/bioinformatics-savvy, so if I find this frustrating what about people who are new to the field?
New Year

To add to the pantheon of great Nerd Shirts out there, we made these T-shirts for the SICB meeting in Charleston, SC which I attended the first weekend of January, along with Christina Zakas and Christine Ewers from my lab. Presumably, this was only funny to the 12 or so members of the symposium I spoke at, but if you understand this: you are also a nerd.
As always, much going on. The RAD-tagged data are back for Notochthamalus, and Christina is working on interpreting these data (hopefully before I give a talk on this project in early March at Auburn!). We also now have specimens of Notochthamalus from Ushuaia, the southernmost city on the planet - no matter what answer their DNA barcodes give up, it will be an interesting one.
We are also pleased to be hosting César Ribeiro, from the Universidade de Aveiro in Portugal. He is a doctoral student with a background in biochemistry and physical modeling, working on the physical oceanography of the Macaronesian Islands and how the oceanography mates with population genetic data. This is not an easy problem, and very similar to what we are trying to do on the east coast of the US and the coast of Chile.
All your base are belong to us
Fecundity 3

Congratulations to Dr. John Robinson, who just finished the easiest Ph.D. defense I’ve yet seen. John came to my lab just less than 5 years ago, and has been a phenomenal addition to our little population here on the third floor and the department as a whole. I won’t, however, miss feeding his Daphnia lines.
This is just a short note between meetings, as we have our final spring faculty meeting in about 15 minutes. I’ve also come to the sad conclusion that BigDyes sequencing reagents do in fact age in the freezer. As my old funding wrapped up and my new funded projects began, I had two full tubes of BigDyes in the freezer, enough to keep us going for a few months - and some new students working on sequencing projects. As the data came back and weren’t very pretty, I thought it might be novice errors, but this week has finally proven that it is time to toss $1800 worth of reagents into the trash.
Technology
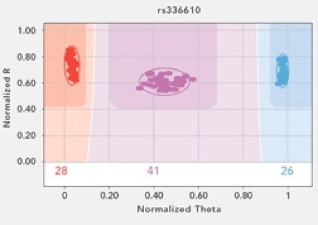
This week has been a good one in the Wares Lab, not least of which because a huge financial gamble has paid off, and is a great indicator of how population genetics and phylogeography is changing. I received some funding from the University of Georgia Research Foundation, intended for junior faculty to generate preliminary data for bigger fish (NSF, NIH) proposals. The entire goal of the award was to try out the Illumina Golden Gate genotyping technology with the Streblospio transcriptome that Christina Zakas is working with. We nervously sorted and re-checked the SNPs in the data, and weeded it out to a set of about 200 that we hoped would be applicable ... and more importantly, scorable. This species has been really recalcitrant when it comes to successful PCR. Fortunately, the Illumina technology doesn’t really go through PCR to get its genotype! In the end, 96 loci were chosen and the array was developed (again, at a big cost for a technology that we’ve never worked with or even seen worked with around here, on a species that we couldn’t even reliably amplify mitochondrial genes).
The data started rolling in this week, and it is phenomenal. Of course some of the loci don’t work well - but around 80% do, which is a better fraction than I’ve ever had when developing primers for nuclear loci in these non-model critters previously. And some DNA templates are not great quality, but close to 95% appear to be just fine. This puts us in position to suddenly be studying these tiny squishy mud worms at a global scale with 2 orders of magnitude more data than ever before. Money well spent, and we’ll be using the same technology on upcoming projects.